
fda lcd monitors for import manufacturer

Today, modern computers displays and televisions (TVs) today use liquid crystal display (LCD), Light-emitting diodes (LED), plasma, or other technologies that do not contain cathode ray tubes (CRTs).
A CRT is a specialized vacuum tube that can be used to receive and display images on an electronic screen. In the early 1960s, some TVs with CRTs were found to emit excessive x-radiation, and a federal performance standard was created to protect the public from this hazard. In the years that followed, the electronic technology for TVs and computer monitors with CRTs changed so drastically that the level of risk of x-ray exposure became almost non-existent. Manufacturers of products that still use CRTs must certify that their products comply with the federal performance standard for the life of the product.
Modern TV receivers and computer monitors provide a benefit for entertainment and information display in many settings. TV receivers and computer monitors containing CRTs no longer pose a risk of emitting any x-radiation. Since the creation of the federal performance standard, the FDA has tested hundreds of TV receivers and computer monitors and rarely encountered any that were unsafe. Most modern computer monitors and televisions (TVs) today use liquid crystal display (LCD), Light-emitting diodes (LED), or plasma and do not contain CRTs or emit x-radiation.
Manufacturers of electronic radiation emitting products sold in the United States are responsible for compliance with the Federal Food, Drug and Cosmetic (FD&C) Act, Chapter V, Subchapter C - Electronic Product Radiation Control.
Manufacturers of televisions and video display products are responsible for compliance with all applicable requirements of Title 21 Code of Federal Regulations (Subchapter J, Radiological Health) Parts 1000 through 1005:
In addition, TV receivers and monitors with CRTs must comply with radiation safety performance standards in Title 21 Code of Federal Regulations (Subchapter J, Radiological Health) Parts 1010 and 1020:

This page will provide an overview of medical and non-medical radiation-emitting electronic products and the requirements that FDA verifies/enforces at the time they are imported or offered for import into United States. To import radiation-emitting products (medical and non-medical) you should first understand what these products are and their requirements.
The Center for Devices and Radiological Health (CDRH) is the FDA center responsible for overseeing the radiation-emitting products program. Visit the Radiation-Emitting Products web page for more information.
FDA defines a radiation-emitting electronic product as any electrically-powered product that can emit any form of radiation on the electromagnetic spectrum. These include a variety of medical and non-medical products such as mammography devices, magnetic resonance imaging (MRI) devices, laser toys, laser pointers, liquid crystal displays (LCDs), and light emitting diodes (LEDs). View examples of radiation-emitting electronic products.
FDA checks the import alert database to ensure the manufacturer or product is not subject to detention without physical exam (DWPE) and listed on an import alert. For example, import alert 95-04 lists certain laser products that fail to comply with applicable performance standards and reporting requirements.
Electronic products are subject to the Electronic Product Radiation Control (EPRC) provisions as defined in the Federal Food Drug and Cosmetic Act (FD&C Act), Chapter 5, Subchapter C, Sections 532 – 538. Radiation-emitting electronic products are regulated by FDA and are required to comply with the general requirements found in 21 CFR 1000-1005. For more information on products subject to performance standards visit the Products Subject to Performance Standards page.
To search for product names and their associated product codes, the radiation type, definition, and applicable performance standards visit the Product Codes for Radiation-Emitting Electronic Products page.
FDA Entry Reviewers are trained to verify compliance with applicable product requirements. The FDA Entry Reviewers use the information provided to FDA in the importer’s entry transmission such as:
These entry declarations are compared to information in FDA’s internal data systems. If the information matches, then compliance is verified; if the information does not match, FDA may gather additional information or may detain the product.
The submission of correct and accurate entry data along with the relevant A of C codes will help expedite the entry review process. Supplying this information accurately increases the likelihood that your shipment will be processed electronically and not held for manual review because FDA’s screening tool, PREDICT, can verify the declared information against FDA internal data systems.
Radiation-emitting electronic products can be considered medical and non-medical products. In addition to the requirements above, radiation-emitting electronic medical products are also subject to medical device regulations. At the time of importation FDA will verify radiation-emitting electronic product requirements (medical and non-medical).
Radiation-emitting electronic products subject to U.S. Federal Performance Standard require submission of Form FDA-2877, Declaration for Imported Electronic Products Subject to Radiation Control Standards, at the time of entry. Products not subject to federal performance standards do not require a Form FDA-2877 for importation into the US. At the time of importation FDA will verify the declarations submitted on Form FDA-2877.
Declaration C – Products that do not comply with performance standards are being held under temporary import bond (TIB); will not be introduced into commerce; will be used under a radiation protection plan; and will be destroyed or exported under CBP supervision when the mission is complete.
Declaration D - Products that do not comply with Performance Standards; are being held and will remain under bond; and will not be introduced into commerce until notification is received from FDA that the products have been brought into compliance in accordance with an FDA approved petition.
The process of “reconditioning” non-compliant radiation-emitting electronic products may be difficult and time consuming which could result in the importer losing the product and money if the reconditioning is unsuccessful. The importer should make a serious effort to understand the FDA radiation safety requirements that are applicable to the products being imported. Reconditioning requests are submitted using the Form FDA-766.
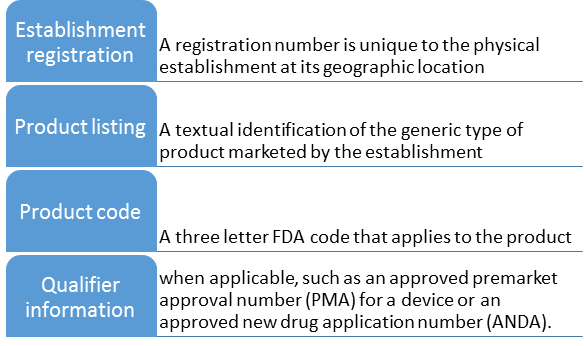
Affirmation of Compliance (A of C) codes are three letter codes that can be provided at the time of import to facilitate FDA review. FDA uses A of C codes to assist in verifying that your product meets the appropriate requirements. Providing the correct A of C code reduces the likelihood that your shipment will be held for further FDA entry review during FDA’s import screening process. Submission of A of C codes is only mandatory in some instances and is not required for all scenarios. Submitting voluntary A of C codes in addition to all mandatory A of C codes may expedite initial screening and review of your entry.
For information on affirmation of compliance codes refer to the “Affirmation Of Compliance References” at the bottom of the affirmation of compliance codes page.

If you are planning on importing any device that is or consists of a tablet or a display screen, take heed of regulatory obstacles that might stand in your way. Since both tablets and display screens often emit radio noise and small amounts of radiation, these devices must comply with Federal Communications Commission (FCC) and the Federal Drug Administration (FDA) standards. In other words, all screen and tablet importers must be certified under both government bodies before importing their products.
The first step, however, is to determine the official classification of your tablet or display screen. The Harmonized Tariff Schedule is the database to find the customs duties that will apply to your product. Below are some tips on how to import your tablet or screen in accordance with FCC and FDA regulations and standards:
Since tablets are distributed for consumer use, they fall under FCC Class B categorization. Tablets and screens are classified as “non-licensed, low-power transmitters,” and fortunately consumers do not need a license from the FCC to use them. However, those importing them will need to get them FCC-certified for safety.
To obtain FCC certification for a product, you need to submit the device for testing at an FCC-authorized lab. Then, the product must be tagged with a compliance label along with an FCC ID (which can be created with a grantee code and a product code). Finally, you can submit the testing results and device specifications, along with the proper labels, to the FCC for approval and they will send you a grant of certification.
Tablets and screens also fall under FDA regulation since they include lasers, LED, or Intense Pulsed Lights, which emit potentially harmful radiation. While the FDA does not technically need to grant approval for products, they do have the right to ban products or order for changes in a device if they do not meet FDA standards. For this reason, importers or manufacturers need to test and certify a product themselves to meet those standards. However, though products are not officially FDA approved, some products – such as medical devices – will require importers to submit reports, which can be done electronically at the FDA’s eSubmitter site. Only after the submission is received will the FDA issue an accession number, which will certify that the product can be marketed commercially.
Importers will also be required to submit a Form FDA 2877 with the accession number and the importing paperwork to Customs and the FDA imports office when their shipment enters the US. As long as the products are labeled and certified properly and the paperwork is in good order, the importing process will go smoothly.

FDA has established electronic product performance standards in its regulations covering several varieties of radiation-emitting electronic products. Companies that manufacture or import electronic products subject to an FDA performance standard are required to submit various electronic product reports, the most basic and essential of which is the Electronic Product Initial Report.
When a manufacturer or importer submits an Electronic Product Initial Report, FDA’s Center for Devices and Radiological Health (CDRH) reviews the electronic product report to ensure the information demonstrates that the manufacturer’s electronic product meets any applicable performance standard and that the process for manufacturing or assembling the electronic product is likely to repeatedly produce complying electronic products. CDRH issues an Accession Numbers to the manufacturer that submitted the electronic product report, and that Accession Number is necessary for importing electronic products into the U.S.
FDA requires importers to provide the Accession Numbers on Form 2877. This is the one assigned by FDA to the manufacturers who made the imported electronic products. FDA import entry reviewers, investigators, and compliance officers use it to confirm that a foreign manufacturer or assembler has at least complied with the most basic FDA regulations governing electronic products: the filing of the electronic product report for the FDA review.
Accession numbers are proprietary information only known to FDA and the company that originally filed the report. As such, there is no public database where you can simply look up the accession numbers for products you are importing. This means that you must ask the manufacturer to reveal their accession numbers to you when FDA demands it upon entry into the United States.
There are also specific certification and labeling requirements that apply to electronic products, and the user manual is a critical piece of information for consumers. FDA requires electronic product user manuals to include many specific warnings and declarations, as well as include clear and concise instructions for use.
FDAImports prepares and files electronic product reports for many different technologies and has former CDRH reviewers ready to assist your company in obtaining new Accession Numbers. We can also help manage the importation process and properly respond to FDA notices or correspondence requests.

This website is using a security service to protect itself from online attacks. The action you just performed triggered the security solution. There are several actions that could trigger this block including submitting a certain word or phrase, a SQL command or malformed data.

This page includes the latest FDA filings for Benq Corp. Currently, you will find the latest 100 filings for Premarket Notifications, Premarket Applications, De Novo Applications, and GUDID registrations.

With more than 195 countries managing imports and exports across the global supply chain, introducing food and beverages into the United States comes with its own unique set of FDA regulations. Understanding and adhering to the regulations for each country can trigger knowledge gaps and spawn uncertainty. Do you need to focus on direct U.S. imports? Employing a holistic import strategy can ensure your goods arrive on time.

A: We can accept TT, Paypal, L/C at sight. 30% deposit and 70% before shipment. We"ll show you the photos of the products and packages before you pay the balance.

This website is using a security service to protect itself from online attacks. The action you just performed triggered the security solution. There are several actions that could trigger this block including submitting a certain word or phrase, a SQL command or malformed data.

This guide serves as an ongoing report of the most recent FDA inspection and enforcement trends, specifically in the area of good manufacturing practice (GMP), based on publicly available data. We"ve included a mix of our firsthand research along with others" analyses and links to the appropriate sources.
Note that the data presented here conform to the fiscal year accounting period for the federal government, which begins on October 1 and ends on September 30. The fiscal year is designated by the calendar year in which it ends.
In November 2022, Jeffrey Meng, program division director, Division of Pharmaceutical Quality Operations III, Office of Regulatory Affairs, stated that the backlog of foreign and domestic onsite inspections for sites considered a high priority. "Looking forward to 2023 and beyond, we have resumed all routine domestic operations and are currently resuming normal foreign inspections. This opening of worldwide operations for FDA will be and is an incredible challenge.” (RAPS)
The number of domestic FDA inspections related to drugs rose from 713 in FY2021 to 756 in FY2022, a ~6% increase. The number of foreign FDA inspections related to drugs rose from 130 in FY2021 to 262 in FY2022, a ~101% increase. (FDA data dashboard)
The number of domestic FDA inspections related to devices rose from 382 in FY2021 to 935 in FY2022, a ~144% increase. The number of foreign FDA inspections related to devices rose from only 4 in FY2021 to 79 in FY2022, a whopping 1875% increase. (FDA data dashboard)
For example, not all inspections are included in the database. Inspections conducted by states, pre-approval inspections, mammography facility inspections, inspections waiting for a final enforcement action, and inspections of nonclinical labs are not included.
#4 —21 CFR 211.100(a) — There shall be written procedures for production and process control designed to assure that the drug products have the identity, strength, quality, and purity they purport or are represented to possess...
#6 — 21 CFR 211.67(b)— Written procedures shall be established and followed for cleaning and maintenance of equipment, including utensils, used in the manufacture, processing, packing, or holding of a drug product...
#7 — 21 CFR 211.25(a) — Each person engaged in the manufacture, processing, packing, or holding of a drug product shall have education, training, and experience, or any combination thereof, to enable that person to perform the assigned functions...
#9 — 21 CFR 211.67(a) — Equipment and utensils shall be cleaned, maintained, and, as appropriate for the nature of the drug, sanitized and/or sterilized at appropriate intervals to prevent malfunctions or contamination that would alter the safety, identity, strength, quality, or purity of the drug product beyond the official or other established requirements.
#10 — 21 CFR 211.110(a) — To assure batch uniformity and integrity of drug products, written procedures shall be established and followed that describe the in-process controls, and tests, or examinations to be conducted on appropriate samples of in-process materials of each batch. Such control procedures shall be established to monitor the output and to validate the performance of those manufacturing processes that may be responsible for causing variability in the characteristics of in-process material and the drug product...
Given that FDA"s inspectional citations specify different descriptions and particular subparts, the interactive chart below breaks down these details to show the relative prevalence of certain observed issues over others.
— Each manufacturer shall establish and maintain procedures to ensure that all purchased or otherwise received product and services conform to specified requirements.
#4 —21 CFR 820.90(a) — Each manufacturer shall establish and maintain procedures to control product that does not conform to specified requirements...
— Each manufacturer shall establish procedures for quality audits and conduct such audits to assure that the quality system is in compliance with the established...
Given that FDA"s inspectional citations specify different descriptions and particular subparts, the interactive chart below breaks down these details to show the relative prevalence of certain observed issues over others.
In 2022, the Office of Pharmaceutical Quality (OPQ) within FDA’s Center for Drug Evaluation and Research (CDER) published its fiscal year 2021 report on the state of pharmaceutical quality. We distilled some of its main takeaways below.
OPQ said its New Inspection Protocol Project (NIPP) program has increased the efficiency of inspections through a more targeted and data-driven approach to identify potential quality problems early on. FDA says its NIPP has “improved how data from pre-approval and surveillance inspections are evaluated and reported.” (FDA has been using these inspection protocols for certain sterile surveillance and pre-approval inspections since 2018.)
Sites making “essential medicines” that protect the public against outbreaks of emerging diseases such as COVID-19 have high median site inspection scores (SIS), indicating a high rate of compliance with GMP. An analysis of active pharmaceutical ingredient (API) and finished dosage form (FDF) sites found that the median SIS for essential medicine manufacturers was 7.45 out of 10, a score that was “significantly higher” than the 7.0 score for non-essential medicine manufacturers. “This observation indicates that sites manufacturing EM products have a higher level of adherence to manufacturing compliance standards than sites that do not manufacture EM products,” OPQ wrote.
For the second year, the number of total recalls (particularly Class I recalls) has increased. This follows a three-year period of declining recalls from FY2017 to FY2019. Since FY2016, recalls spiked up dramatically, going from roughly 300 recalls events a year in 2019 to 700 in FY2020 to 800 in FY2021. Hand sanitizers that contained methanol, as well as consumer products and sunscreens with benzene contamination are largely to blame for the increase.
Roughly half (49.1%) of 1,143 eligible firms did not submit field alert reports (FARs) to the agency over a four-year period from FY2018 to FY2021. FDA’s postmarket reporting requirements specify that sites submit FARs after receiving information on significant quality problems with their distributed drug products.
A growing number of products are failing sampling and testing requirements; a method of inspection is used when FDA cannot get to sites to conduct inspections. In FY2021, the percentage of non-compliant samples grew to 35%, an increase from 16% in FY2020. The growing rate of non-compliance “is driven by focused sampling assignments with high non-compliant rates for products with nitrosamine contamination, hand sanitizers, and sampling related to COVID-19 mission critical sampling and testing, which became more prominent in FY2021.“
Read FDA"s full FY2022 report (PDF) on fda.govhere. We distilled some of its main takeaways below. Read our blog post for more depth into some of these takeaways.
FDA used Mutual Recognition Agreements (MRAs) and its authority to obtain records for sites in advance or in lieu of inspections due to the pandemic.In 2012, the Food and Drug Administration Safety and Innovation Act gave FDA new authorities under the Food, Drug, and Cosmetic Act §704(a)(4) to request records or other information from firms in advance of or in lieu of an inspection.
“FDA surveillance history, requests for records, and inspection reports obtained through the MRAs were all used to mitigate risk and enable regulatory actions.”
“MRA authority was used to assess 183 sites through MRA inspection reports for a total of 745 sites (18% of the FY2020 CDER Site Catalog). For comparison, in FY2019 1,258 drug quality assurance inspections were performed and an additional 109 sites were assessed using MRAs for a total of 1,367 sites (32% of the FY2020 CDER Site Catalog).”
“As in past years, the majority of Warning Letters in FY2020 were issued to sites with non-application products (69%), and especially those that manufacture finished dosage form (FDF), non-sterile, non-application products (41% of all Warning Letters).”
“Import Alerts doubled to 128 in FY2020. Latin America had the most sites on Import Alert for the first time in FY2020, due to an unprecedented number of new hand sanitizer registrants from Mexico that failed to meet quality standards."
Since FY2019, there has been a small decrease (0.10) in the mean Site Inspection Score (SIS) of the entire inventory of sites (7.3).FDA’s SIS, a scale of 1 to 10, is used as a proxy for compliance with CGMP regulations. The SIS is based on the classification of FDA drug quality assurance inspections conducted over the prior ten years, including inspections classified under the MRA program, which allows some global regulators to recognize reports from their counterparts’ inspections.
“For FY2016–FY2020, three defect categories account for 60% of all defects reported: Product Quality Questioned, Device Issues, and Packaging Issues.”
“The most substantial increases in the number of recalls by industry sector in FY2020 were in the No Application and NDA & ANDA (i.e., sites manufacturing for both application types) sectors.” (See full report for additional details.)
“Each major recall over the last five fiscal years was associated with microbial or chemical contamination/impurities; a focus area for the industry to improve quality.”
CDER will continue to seek to minimize long-standing problems such as drug shortages due to quality issues through proactive efforts including the New Inspection Protocol Project (NIPP) and Quality Management Maturity. The NIPP “is aimed at using standardized electronic inspection protocols to collect data in a structured manner. The protocols promote consistent and comprehensive coverage of critical areas of drug manufacturing and provide structured, data-rich reports.
“In the future, FDA will have the ability to better understand how certain variables (e.g., location of the establishment, type of establishment) affect quality. As more data are collected through NIPP, these types of insights can inform future inspections, identify policy/outreach opportunities, and influence application-related decision making."
A report published by the ECA Academy looked back from October 2018 to September 2019 to review the FDA warning letter trends among pharmaceutical manufacturers.
Where applicable, we’ve provided links to relevant resources and next steps for those looking for guidance and assistance in mitigating trending risks.
The specific issues contained within these recent warning letters reveal a continuation of a trend that’s been running for years: lapses in meeting basic GMP requirements.
As analyzed and compiled by Barbara Unger in an impressively researched column for Pharmaceutical Online, the frequency of Forms 483 issued to pharmaceutical companies has continued its steady rise over the past few years.
Note that for this and the following sections covering inspection observation trends, the data presented adheres to the analysis methodology and limitations described in the introduction of the
Again, given the caveat of the limited public data available (the FDA’s data includes only Forms 483 issued through its electronic system and omits API manufacturers), some notable findings emerge regarding specific §211 citations.
As we explored in another article, 2019 saw the FDA put a greater compliance focus on over-the-counter (OTC) drugs and other health product manufacturers. In a July 2019 column for Pharmaceutical Online, we analyzed these OTC-specific compliance trends, pulling out the following common issues appearing in warning letters.
Nonconformance Management —Another recent trend afflicting OTC drugmakers mirrors a broader and well-documented trend throughout the drug and device space: inadequate nonconformance management. As demonstrated by a large number of citations issued specifically for “inadequate, incomplete, and undocumented investigations,” these warning letters offer evidence of a long-standing perception that an outsized focus is placed on immediate nonconformance correction rather than on thoroughly investigating and executing corrective and preventive actions following a comprehensive root cause analysis.
Roles, Responsibilities, and Authority of the Quality Unit —The internal quality unit (QU) has been the target of many recent warning letters to OTC drug and health product manufacturers as an underlying cause of product quality and GMP compliance problems. Numerous firms have been cited for having an inadequate QU. In the most egregious examples, firms lacked this designated team entirely. More often, however, regulators have cited firms for a lack of written procedures that govern the responsibilities and functions of this group. 21 CFR Part 211 is clear about the need to establish a “quality control unit” with the documented responsibility and authority to make critical decisions.
Based on a growing number of relevant warning letters, as well as analyses of enforcement trend data and public statements made by the FDA, it’s clear that a renewed focus has been placed on evaluating manufacturers of OTC drug and health products in key areas of GMP.
These areas of enforcement focus include ineffective quality units, poor testing of incoming materials and components (i.e., relying on a supplier’s certificate of analysis), poor product testing, poor analytical and microbial testing and validation methodology (including method suitability), and inadequate nonconformance management.
Here are the major takeaways for inspections and compliance, specifically back from FDA"s Report on the State of Pharmaceutical Quality: Fiscal Year 2019 :
Based on its 10-point inspection score, the overall average for inspections in FY2019 was 7.4.(US- and EU-based sites scored averages that were slightly higher: 7.7 and 7.6, respectively.)
Based on its 10-point inspection score, homeopathic products and sterile over-the-counter (OTC) products had the lowest average scores at 6.5 and 6.2, respectively. For more on recent chronic quality and compliance issues among OTC and health product manufacturers,
In its report, regulators went on to say, "These sections represent some of the key elements of an effective Pharmaceutical Quality System. These are potential areas of focus for manufacturing facility management to improve overall pharmaceutical quality and inspectional outcomes."
Publicly available warning letters and inspection observation data provide powerful resources for understanding areas of regulatory focus and a benchmark for evaluating potential vulnerabilities within the quality system and beyond.
In many of its warning letters, the FDA has “strongly recommended” engaging a third-party consultant qualified in the relevant regulations to assist in meeting CGMP requirements.
While we help many companies resolve Forms 483 and warning letters, we also help prevent them from being issued in the first place. At The FDA Group, we plan and conduct effective internal quality audits to ensure your quality system is completely aligned with all documentation and operations — the critical part of any internal audit.

Millions of parents rely on our formula to feed their babies. And we know that our recent recall caused additional stress and anxiety in an already challenging situation of a global supply shortage. We are working hard to help moms, dads and caregivers get the high-quality nutrition they need for their babies.
Abbott is working closely with the U.S. Food and Drug Administration (FDA) to restart operations at the Sturgis, Mich., facility. We continue to make progress on corrective actions and will be implementing additional actions as we work toward addressing items related to the recent recall. In the meantime, we are working to increase the supply of infant formula by prioritizing infant formula production at our facilities that provide product to the U.S. market.
We have an FDA-registered plant in Cootehill, Ireland, where we"ve increased the volume of Similac Advance powder formula produced for the U.S. We"re air-shipping product from this facility into the U.S. daily and the product is being restocked regularly.
Our Cootehill team sources ingredients from approximately 1,000 dairy farms in the local area. Following stringent quality and safety processes, each batch of infant formula undergoes extensive quality checks before it reaches stores.
Columbus, Ohio, is the headquarters for Abbott"s U.S. nutrition business and is home to one of our five manufacturing facilities that produce infant formula for the U.S. market. At this facility we"ve made significant changes to ensure we can prioritize production of Similac Ready-to-Feed liquid formula, a product that can be used directly from the bottle. In the second quarter of the year, we expect to produce nearly three times more Similac Ready-to-Feed liquid formula than we did during the same period of time last year. And this product will be available on retail shelves and online soon.
Across the U.S., we"re prioritizing production of infant formula products to help replenish the supply in the market. And, this year, we will more than double the amount of Similac Advance powder formula we"re bringing in from our manufacturing facility in Cootehill, Ireland.
We are dedicated to doing everything possible to ensure parents and caregivers have what they need to feed their babies. And we"re always focused on what we can do to continue to serve our customers. We will continue to work closely with the FDA to implement corrective actions at the Michigan facility.
We are currently reviewing the FDA"s observations as provided in its Form 483 from its inspection of our powder formula manufacturing in our Sturgis, Mich., facility. We"re taking this very seriously and are working closely with the FDA to implement corrective actions.
While there are actions we need to take to address the FDA observations, it is important to note that no Cronobacter sakazakii or Salmonella was found in any of our testing of products distributed to consumers. Additionally, the unique genetic makeup of the Cronobacter sakazakii microbes found in non-product contact areas at the Sturgis facility did not match the Cronobacter sakazakii microbes from the reported cases. This follows the FDA"s removal of the Salmonella case from its investigation earlier this month.
Abbott is committed to upholding the highest standards for manufacturing of all nutrition products. We have already begun implementing corrective actions and enhancements at the facility, leveraging new technology and strengthening our processes, to give parents and customers renewed confidence in the quality of manufacturing at our Sturgis plant when we restart operations there. Our actions include:
Consulting with industry experts to implement the latest technological advancements in food manufacturing processes, including a 3D augmented reality system, which will provide a clearer visualization of product as it moves through the facility, enhancing Abbott’s ability to make informed decisions (including remotely) during the manufacturing process
We know there are constraints in infant formula supply and we’re taking action to help address this. We have a global manufacturing and supply network we’re leveraging to better meet demand and increased production at an FDA-registered facility in Europe and are air freighting in Similac Advance infant formula powder. Our other U.S. plants continue to supply infant formula to the market and we’re prioritizing some production from other liquid nutritional products to Similac. Actions we’re taking to address supply include:
Working with USDA and state agencies to provide authorization for parents who get formula from the Special Supplemental Nutrition Program for Women, Infants and Children (WIC) with other Similac products at no cost, including for other manufacturers" products
We know that millions of parents and caregivers around the world count on our formulas to feed their babies and children and we are doing everything possible to address this situation.

This website is using a security service to protect itself from online attacks. The action you just performed triggered the security solution. There are several actions that could trigger this block including submitting a certain word or phrase, a SQL command or malformed data.

With more than 195 countries managing imports and exports across the global supply chain, introducing food and beverages into the United States comes with its own unique set of FDA regulations. Understanding and adhering to the regulations for each country can trigger knowledge gaps and spawn uncertainty. Do you need to focus on direct U.S. imports? Employing a holistic import strategy can ensure your goods arrive on time.
Affirmation of Compliance (A of C) codes are three letter codes that can be provided at the time of import to facilitate FDA review. FDA uses A of C codes to assist in verifying that your product meets the appropriate requirements. Providing the correct A of C code reduces the likelihood that your shipment will be held for further FDA entry review during FDA’s import screening process. Submission of A of C codes is only mandatory in some instances and is not required for all scenarios. Submitting voluntary A of C codes in addition to all mandatory A of C codes may expedite initial screening and review of your entry.
FDA evaluates product compliance and manufacturer quality control and testing programs by reviewing reports as needed. Depending on reporting requirements, manufacturers may be required to submit reports to FDA that describe the manufacturer, the product, its radiation safety specifications, any product labeling, and the quality control testing program in place used to ensure compliance.
In plain language, a product report tells the FDA how the product complies with the performance standard. The report must be complete and include detailed responses to all requests for information. Part 21 CFR 1002 contains specific reporting and recordkeeping requirements for product, supplemental, and abbreviated reports.
Many manufacturers are also required to submit annual reports to the FDA. These describe annual production levels, summary results of required compliance testing, and a summary of radiation concerns and user safety inquiries received over the year. The annual report is due each September 1st, for the prior reporting period of July 1 through June 30. An example is listed on this slide. The annual report is valid for one year.
There are three ways to submit your report to the FDA. First, you may prepare an electronic copy and submit it electronically. Second, you may prepare an electronic copy and mail that to us. And finally, you may prepare a hard copy and mail that. I"ll review each method, including the benefits of some, so you may choose the method you prefer to use.
Foreign Supplier Program ViolationsThe FDA continues to issue warning letters to importers concerning noncompliance with requirements under the Foreign Supplier Verification Program, which requires importers to perform certain risk-based activities to verify that the human and/or animal food they import has been produced in a manner that meets applicable U.S. food safety standards.
Most recently, FDA inspections of the facilities of one importer found that it failed to develop, maintain, and follow an FSVP for fresh enoki mushrooms. The FDA has given the importer 15 working days to provide information on the specific things it is doing to correct this violation; e.g., documentation of changes made and records to demonstrate implementation of an FSVP. If the importer does not act promptly the FDA may take further action, such as refusing admission of violative products and subjecting the importer to detention without physical examination.
The FDA notes that the mushrooms at issue have already been placed under two import alerts and that this information should be taken into consideration when the importer evaluates its suppliers and develops its FSVP.
Import Alerts: Cookware, Medical Instruments, Seafood, ProduceFDAimport alertsaffecting the following have been newly issued or modified in the past week.
Import alerts inform FDA field staff that the agency has enough evidence or other information to allow a product that appears to be in violation of FDA laws and regulations to be detained without physical examination at the time of entry. Import alerts may cover products from designated countries or areas (including from all foreign countries), manufacturers, or shippers.
Firms and/or products on the “red list” of an import alert are subject to Detention without Physical Exam (DWPE), while firms and/or products on the “green list” are not because they have met the criteria for exclusion. Some import alerts include a “yellow list” of firms, products, and/or countries subject to intensified surveillance because the nature of the violations may warrant further field examinations of individual entries and/or additional analyses. In addition, depending on the specific import alert, shipments of products subject to DWPE may still be imported into the U.S. if the importer has demonstrated that the shipment is in compliance.
If a product is detained without physical examination the importer has the right to provide evidence to the FDA in an attempt to overcome the appearance of the violation. If no such evidence is submitted, or if the evidence provided is insufficient, the product will be subject to refusal of entry into the U.S.



 Ms.Josey
Ms.Josey 
 Ms.Josey
Ms.Josey