
multiplexing lcd displays free sample
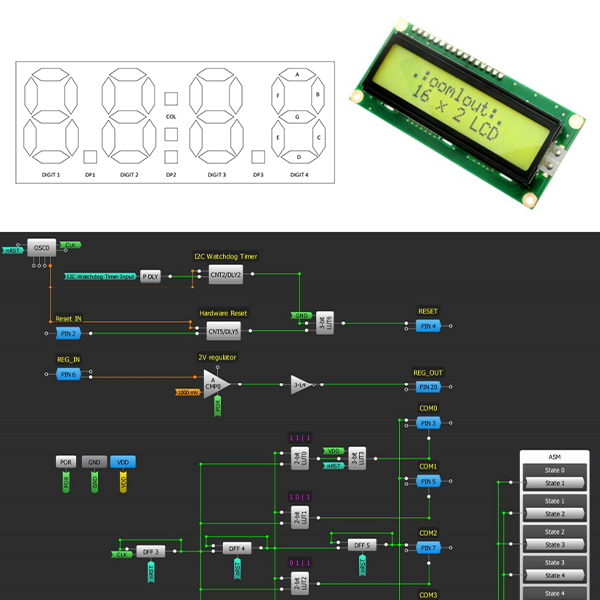
Driving a segment LCD using multiplexing reduces the number of pins required to turn on or off the segments of a display. This application note discusses what a segment display is and the driving method in detail. The SDAF102NCRN01, a 3V, 7-segment custom LCD is used as an example.
Segment LCDs are constructed using two pieces of Indium Tin Oxide (ITO) glass with a twisted nematic fluid sandwiched in between. The majority of these displays are custom-made. Typical applications include measuring acidity levels in swimming pools, gases, or temperature. There are two types: static and multiplexed. A static display is a segment display with one pin for every one segment, whereas a multiplexed LCD has grouped segments, reducing the number of pins.
So, what is a segment? A segment is any line, dot or symbol that can be turned on and off independently. The figure below shows an example of a custom display developed by Focus LCDs.
The number of segments is dependent on what will be displayed. The most popular are seven-segment displays. In Figure 1, the digits: “0”, “8”, “4”, “7” are all seven segments. Each segment can be independently turn on or off to show a letters or number. However, the range of letters are limited. While fourteen segments have the ability to display any number and more letters.
Icons such as symbols for battery, signal strength, plus/minus and bar graphs are also considered segments. Finally, segments can also be “permanent”. This means it is always on even with no power. This is accomplished by burning the segment onto the glass. The text: “FOCUSLCDS.COM” in Figure 1 is an example.
The multiplexing technique aims to reduce the number of pins that are necessary for driving the segments of the display. This results in a simplified LCD module. In this setup, each segment control line can be connected to as many segments as there are backplanes, provided that each of the connected segments are tied to separate backplanes. This method "multiplexes" each of the segment control lines and minimizes the number of pins. The advantage of this is increased display density and reliability at the expense of complicated drive circuitry.
Segments are turned on or off using an AC voltage with no DC component (Figure 4). This means that the average voltage of the AC waveform should be equal to zero. Having a DC bias will reduce the life of the display. Also, there must be always an AC voltage on all the segments of the LCD. A sign that the LCD is degrading is when there is a loss of alignment on the edge of the characters, resulting in a distorted visual appearance.
Crosstalk, or “ghosting”, occurs when an LCD is overdriven by a combination of frequency and voltage. This appears as a partial turning on or off of a segment. To prevent inadvertent turning on or off of the segments, unused segments must be connected to its backplane (COM) pins.
One major disadvantage of multiplexed drivers is reduced contrast due to a lower duty cycle. In this case, a segment is on 25% of the time, while in static-driven displays have sharper contrast from being on 100% of the time. However, to the human eye this decrease in contrast is not noticeable.
Buyers and others who are developing systems that incorporate FocusLCDs products (collectively, “Designers”) understand and agree that Designers remain responsible for using their independent analysis, evaluation and judgment in designing their applications and that Designers have full and exclusive responsibility to assure the safety of Designers" applications and compliance of their applications (and of all FocusLCDs products used in or for Designers’ applications) with all applicable regulations, laws and other applicable requirements.
Designer agrees that prior to using or distributing any applications that include FocusLCDs products, Designer will thoroughly test such applications and the functionality of such FocusLCDs products as used in such applications.
1TN (Twisted Nematic) is a popular LCD technology that does not require current flow for TN cells to work and uses lower operating voltages, making them suitable for portable applications.
Unbundled Channelization (Multiplexing) 5.7.1 To the extent NewPhone is purchasing DS1 or DS3 or STS-1 Dedicated Transport pursuant to this Agreement, Unbundled Channelization (UC) provides the optional multiplexing capability that will allow a DS1 (1.544 Mbps) or DS3 (44.736 Mbps) or STS-1 (51.84 Mbps) Network Elements to be multiplexed or channelized at a BellSouth central office. Channelization can be accomplished through the use of a multiplexer or a digital cross-connect system at the discretion of BellSouth. Once UC has been installed, NewPhone may request channel activation on a channelized facility and BellSouth shall connect the requested facilities via COCIs. The COCI must be compatible with the lower capacity facility and ordered with the lower capacity facility. This service is available as defined in NECA 4.
Sample multiplexing is a technique in proteomics that has been introduced in 1999, enabling scientists to compare and analyze two different sample preparations simultaneously within a single MS injection.3 scan. Sample multiplexing in proteomics increases the confidence of results due to the ability to compare and analyze high numbers of biological samples in tandem while also including an internal standard or quality control for normalization.
In this review, a focus on approaches that increase sample throughput in quantitative proteomics with sample multiplexing is provided. Sample multiplexing strategies for proteins
One of the main advantages of sample multiplexing is the ability to combine multiple samples and analyze them in a single MS run, reducing the instrument time and cost. There are multiple methods used for relative quantification, which are a consistent way to analyze protein expression patterns and the effects of biological perturbation in disease states. Quantitative proteomics is performed either by label-free methods or by isotopically labeling proteins or peptides prior to MS analysis. Isotope incorporation can be performed at the protein or peptide level using, for example, 2H, 13C, 15N, or 18O, as heavy isotopes.13C, 15N, or 18O isotopes which do not alter the chromatographic interaction of the analyte when combined. While the samples are pooled and analyzed, similar peptides from different samples elute at the same m/z in the MS scan with similar retention times.
Multiplexing capabilities for combined analysis of more than eight proteomes. (A) Structure of tandem mass tags with the table showing the number of possible isotopes for 6-plex, 8-plex, 10-plex, and 11-plex reagents. (B) Structure of DiLeu and its isotopomers. (C) Structure of iTRAQ with its isotopomers.
Increasing the multiplexing capacity of the stable isotopes and isobaric tags helps us to analyze similar peptides from different samples in a single experiment. As mentioned, this increases the sample complexity while the presence of coeluting ions of similar charge can affect the accuracy and precision of the quantitative label using isobaric tags.3 spectrum instead of the MS/MS spectrum.3 isolation, however, is that it results in fewer proteins quantified.
The ability to multiplex utilizing isobaric tags either stand-alone or in combination with metabolic or chemical tags provides an opportunity for unbiased biological discovery. Approaches which multiplex up to 54 samples in a single MS injection represent a drastic increase in throughput, reproducibility, and robustness of quantitative proteomics. Multiplexing approaches currently available have developed tremendously in recent years, with a capability to process and compare up to 12 samples in a single injection. Proof-of-concept studies have been reported to combine 24–54 samples
In 2012, Noah and Gygi showed that multiplexing can be increased up to 18 samples in a single experiment. Multiplexing was demonstrated by combining triplex metabolic labeling and 6-plex isobaric tags using three separable MS SILAC metabolic labels and corresponding TMT 6-plex isobaric tags (3 × 6) (Figure 3A).Figure 3B). The novel medium tag is 71.02 Da (C3H5NO) heavier than the light tag, while the heavy tag is 14.02 Da (CH2) heavier than the medium tag. The caveat is that the three TMT tags elute at different times from the column. The proof-of-concept experiment showed the multiplexing ability by combining 54 experimental conditions to one sample using a kinase activity assay (Figure 4). The figure shows the inhibition of protein kinase A (PKA) in solution (Figure 4A) and breast cancer cell line MCF7 lysate by a peptide inhibitor (PKI) at 18 different conditions analyzed with triplicate injections (Figure 4B). A single peptide (residue 243–252) from the PKA substrate (EPB42) was serine phosphorylated upon inhibition (Figure 4C). The study further compares three different peptide transitions of the PKA substrate peptides by SILAC labeling and six variations from TMT, resulting in nine peptide transitions in a single study. The TMT and SILAC labeled peptides elute at different times, with the light eluting earlier than medium and heavy labeled peptides (Figure 4D). The peptides were quantified throughout the three different transitions with ultimately low standard error (8.75%). The increase in sample multiplexing helps researchers to reduce the instrument time and sample handling, while the novel TMT tags are not commercially available. This makes it difficult to access by the global research community.
Combination of metabolically and chemically labeled stable isotopes to increase the sample multiplexing capabilities using a triplex SILAC and 6-plex TMT simultaneously. (A) Metabolic labels provide intact mass differences distinguishable in an MS1 scan of intact peptide ions. Upon isolation and fragmentation of the light, medium, and heavy versions of a peptide, the isobaric labels provide separate multiplexed quantitative measurements for each in the MS/MS spectra. (B) Structure of the novel TMT tags. Reproduced with permission from
Increasing the multiplexing capabilities of tandem mass tags for combined analysis. Two 54-plex analyses consisting of an 18-point IC50 curve performed in triplicate. Inhibition of PKA using the peptide inhibitor PKI was conducted in solution (A) using 2 ng of commercially available PKA and in 5 μg of lysate from the breast cancer cell line MCF7. (B) Three variants of the substrate peptide based on two novel TMT reagents (C), resulting in nine target peptides. The resulting three variants eluted at different retention times (D). Reproduced with permission from
Isobaric tags in combination with dimethyl tags or metabolic tags like SILAC help enhance the multiplexing ability of the isobaric tags. Various studies have successfully enhanced the multiplexing ability using dimethyl tags and SILAC in combination with TMT, iTRAQ, and DiLeu. Global measurement of protein turnover in primary human dermal fibroblasts has been shown as a proof-of-concept for enhanced multiplexing using SILAC and TMT labeling.
Enhanced multiplexing of cPILOT is achieved by combining precursor MS labeling with isobaric tags (TMT, DiLeu) or iTRAQ. cPILOT uses stable isotope dimethylation of peptide N-termini with light [−(CH3)2] and heavy [−(13C2H3)2] isotopes at low pH (~2.5), which keeps the lysine residue available for subsequent high pH (8.5) TMT tagging.
Enhanced multiplexing capabilities of 12-plex cPILOT for global proteome approach using a 6-plex TMT. Proteins from three different tissues of wild-type (WT) and Alzheimer’s disease (AD) mice were extracted, reduced, and alkylated and randomly grouped into two groups, dimethylated with light and heavy isotopes, tagged with TMT 6-plex reagents, and injected to an LC-MS/MS system. Precursor data shows light and heavy dimethylated peptides, represented by the peaks at m/z 643.854 and 647.875. These peptides were selected, isolated, and fragmented, thus generating CID-MS/MS spectra, which provided peptide identification. An additional isolation and fragmentation of the most intense fragment ion of the light and heavy dimethylated peptides generated HCD-MS3 spectra, respectively. The peptide sequence is T(dimethyl)ELNYFAK(TMT6) and belongs to phosphoglycerate kinase 1. Reproduced with permission from
Enhancing the multiplexing capabilities of sample preparation using isobaric tags in combination with cPILOT. Spectra acquired on the Orbitrap Fusion Lumos of a DiLeu cPILOT-labeled peptide with sequence DSILEVLK. The coeluting light and heavy peptide peak pair, with overlapping extracted ion chromatograms (XIC), is detected in the MS scan at m/z 545.3383 and 549.3597, and each is acquired by CID MS/MS in the linear ion trap. Following each MS/MS scan, HCD SPS-MS3 acquisition in the Orbitrap (RP 60K) of the top four ions (marked by gray asterisks) generates two sets of abundant 12-plex DiLeu reporter ions for 24-plex quantification. Reproduced with permission from
The key issues to be taken care of while performing a cPILOT analysis include careful handling of samples, exposure of similar reaction times to all samples, and maintaining low pH for dimethylation. Sample handling is very important in cPILOT to make the mixing of light and heavy samples similar in each analysis. For large numbers of samples (10s-100s), this is best facilitated by using a multichannel pipet, processing in batches, or using a robotic platform. The dimethylation reaction of cPILOT is pH specific and depends on maintaining low pH for dimethylation to prevent labeling of lysine residues. While processing large numbers of samples is desirable, the major requirement is to quantify dimethylated pairs from all of the reporter ion channels (11–12 plex). This can be achieved by using current state-of-the-art mass spectrometers (Orbitrap Fusion or Lumos) and nano flow LC systems. Additionally, it is advisable to optimize the parameters such as LC gradient time, m/z isolation window, dynamic exclusion time, targeted analysis nodes, selective y1- fragmentation,3 for obtaining maximum multiplexing capability and analytical performance.
Sample multiplexing helps researchers to study proteome changes throughout multiple cohorts so as to study the effects in healthy and disease conditions. With the ability to combine up to 54 samples in a single study, it will be easier to compare and study the interaction, modification, and pharmacokinetics of a protein/molecule at one glance. Sample multiplexing drastically reduces the time required for sample analysis.
Samples for proteomics studies are mostly tissue/clinical materials from patients for discovery and validation of biomarkers. Samples are biological fluids such as plasma, cerebrospinal fluid, urine, saliva, and tissues from human, animals, cell lines, and plants. There are two main limitations which have to be overcome to make use of enhanced multiplexing strategies at its full efficiency for such samples. The first limiting factor is sample multiplexing reagents and the other factor is analytical instruments used to prepare and analyze the samples.
Molecular medicine is moving beyond genomics to clinical proteomics which will require robust methodologies for biomarker profiling. Proteomics plays a crucial role in early diagnosis, prognosis, and disease monitoring, and there is a need to analyze hundreds to thousands of samples to compare the expression of proteins in multiple experimental conditions. Protocols that are amendable to automated sample preparation will be necessary because handling 1000s of samples manually will require weeks to months to prepare samples, while analysis is possible within a single or a few days. The main problem faced by academic researchers is the expensive costs of the multiplexing reagents and automated platforms. Use of multiplexing strategies that offer >8–54 sample multiplexing, increase the potential of achieving large-scale initiatives in a high-throughput manner.
Albert B. Arul studied Biochemistry at the University of Madras (India), where he received his Masters and Ph.D. degrees in 2002 and 2009, respectively. After a short stay at Vimta Laboratories Ltd for a year at Hyderabad, India, as a Scientist B at a preclinical drug discovery group, he moved to the college of Applied Medical Sciences, King Saud University, Kingdom of Saudi Arabia (2010–2012). Later, he moved to South Korea as a Research Assistant Professor to work on automated sample preparation for biomarker discovery using proteomics from 2012 to 2016 at Gachon University and Seoul National University. After completing postdoctoral training at South Korea, he moved to George Washington University, Washington, DC, as a Postdoctoral scientist to work on identification of MoA of drug molecules using quantitative proteomics. Currently, he is working as a Research Assistant Professor at the Department of Chemistry, Vanderbilt University. His current research focuses on developing sample multiplexing strategies and automation of the sample preparation for proteomics studies.



 Ms.Josey
Ms.Josey 
 Ms.Josey
Ms.Josey